Kmer Database Format (Part 1.)
Review of K-mers and Motivation for .kdb

The goal of this blog post is to introduce the concepts of k-mer subsequences and blocked GNU-zip file (.bgzf) and suggest that they be used together to form a new file specification for younger bioinformaticians. If I'm successful, the reader should have a basic understanding of common k-mer packages, my opinion on the algorithms and APIs, and the challenge of understanding advanced computer science and benchmarking concepts utilized by those packages/algorithms from the eyes of novice, beginner, and even intermediate bioinformatics students.
The software concept I'm introducing here does not have a name, specification, or even a prototype. The target audience of this blog post is biologist-turned-bioinformaticians that might want an example of a class project that doesn't revolve around human genomics or alignment algorithms. If you like the concept of this post being at the intersection of algorithm design, academics, and Illumina-hype then give this blog a share, retweet, upvote, or some Reddit silver when I post in on /r/bioinformatics.
~7k words. About a 25-35 minute read
What are K-mers?
K-mer spectra are like absorbance spectra
K-mers are short (10-30bp) subsequences of larger biological sequences with some choice of subsequence length referred to as k. Biologists are first introduced to the k-mer problem as the minimum specificity above 'random' or 'background' necessary for the design of primers, in the 18-25bp range.
The K-mer problem Welcome to the Kompany is a fundamental problem in bioinformatics that has led to advancements in genomic problem solving techniques and usable results from the Human Genome Project
Welcome to the Kompany is a fundamental problem in bioinformatics that has led to advancements in genomic problem solving techniques and usable results from the Human Genome Project Because thats how you get anterior evodevo network topologies assembly
Because thats how you get anterior evodevo network topologies assembly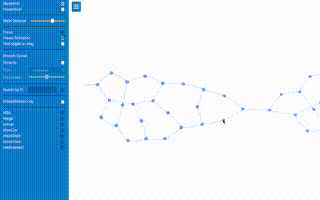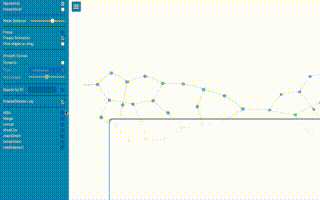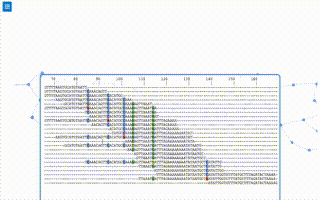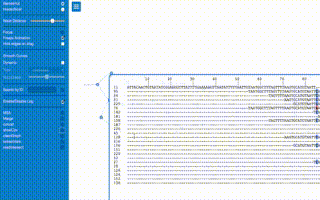 coelias/Pynteractive and statistical analyses
coelias/Pynteractive and statistical analyses problem_solving_is_not_math.gif.
problem_solving_is_not_math.gif.
Classically fight_fire_with_fire.gif, k-mers have seen the most use in overlap-layout-consensus
fight_fire_with_fire.gif, k-mers have seen the most use in overlap-layout-consensus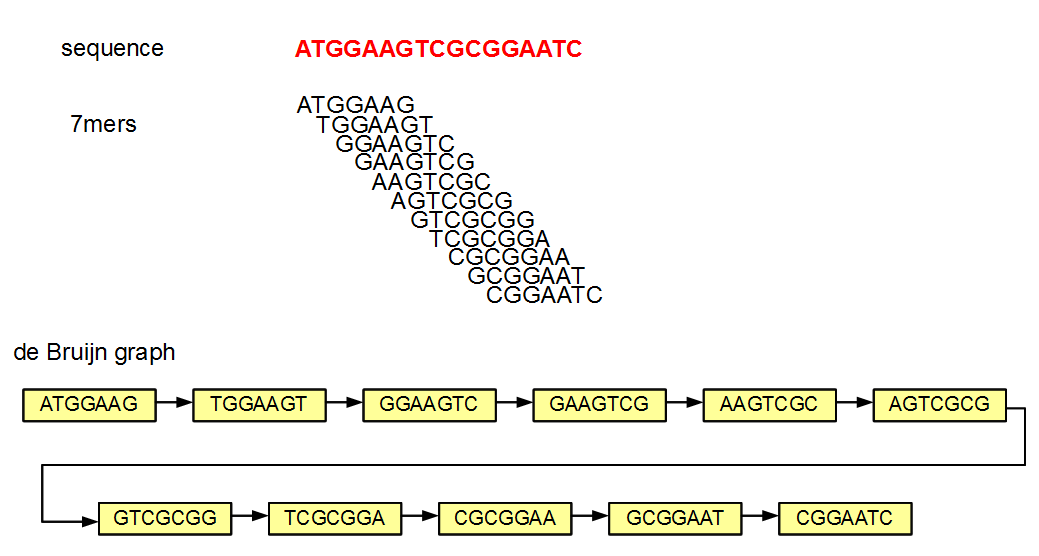 How a de Bruijn graph is built from k-mers assembly methods. With some simplification of the problem and math, the k-mer problem can be stated as "how much overlap is statistically significant to use as a consistent subsequence size to unite the sequence reads to form contigs" and was the critical theoretical bottleneck after the BACs were sequenced in the human genome project.
How a de Bruijn graph is built from k-mers assembly methods. With some simplification of the problem and math, the k-mer problem can be stated as "how much overlap is statistically significant to use as a consistent subsequence size to unite the sequence reads to form contigs" and was the critical theoretical bottleneck after the BACs were sequenced in the human genome project.
The problem is also featured in phylogeny Midichlorian community evolution due to domineering theoretical emphasis on local alignment
Midichlorian community evolution due to domineering theoretical emphasis on local alignment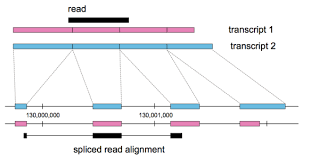 Calculating abundances of overlapping transcripts by splitting read-counts across genomic regions vs separate transcripts. Aligning reads to genomes can capture unknown transcripts better by focusing efforts on reads that align to inter-genic regions, regions thought to be unexpressed according to the current annotations., the moving targets of both classification and distance metrics, and the lack of reference sequences in non-model species. The problem was also used in pseudoalignment
Calculating abundances of overlapping transcripts by splitting read-counts across genomic regions vs separate transcripts. Aligning reads to genomes can capture unknown transcripts better by focusing efforts on reads that align to inter-genic regions, regions thought to be unexpressed according to the current annotations., the moving targets of both classification and distance metrics, and the lack of reference sequences in non-model species. The problem was also used in pseudoalignment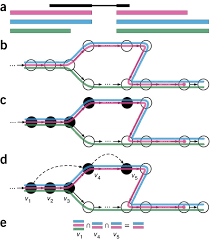 In a) the read (black) shares overlap with three transcripts. b) a graph represents the differences between the exon boundaries of the transcripts c) reads mapped onto that graph d) arrows overlaying splice site inforation e) set notation describing the transcripts the read most likely originated from methods in contrast to highly-engineered alignment algorithms like Bowtie and BWA.
In a) the read (black) shares overlap with three transcripts. b) a graph represents the differences between the exon boundaries of the transcripts c) reads mapped onto that graph d) arrows overlaying splice site inforation e) set notation describing the transcripts the read most likely originated from methods in contrast to highly-engineered alignment algorithms like Bowtie and BWA.
How did you learn about this problem?
Every student wonders "what about 24bp makes it sufficient for my first RT-PCR experiment?"
Dr. Viswanathan posed this question after I mentioned interest in mathematical biology. Why do nearly all biologists choose primers of that length; what is the magic behind the number? Though I struggled to articulate my questions, they remained. Was the choice sufficient for the transcribed or coding regions only? Both sides of the genome? Did we need to BLAST the primers against the genome for secondary hits? All of those questions and their directions turn basic assumptions about 'necessary' specificity into derivations about mathematical biology; and their application areas included miRNAs, RNAi, CRISPR, and multiplexed expression quality control. I was hooked.
Decisions in the Face of Assembly or Just Enough Statistics to be Dangerous
A genome, fasta, fastq can be sliced into uniform subsequences and tallied. The resultant vector, which can be sparse A sparse matrix or vector is one that has abundant zeroes in it. In this case, sparseness comes from unobserved k-mers, with count of 0, referred to as nullomers., is the k-mer profile. Thus, specificity/resolution in k-mer space requires additional space in memory.
A sparse matrix or vector is one that has abundant zeroes in it. In this case, sparseness comes from unobserved k-mers, with count of 0, referred to as nullomers., is the k-mer profile. Thus, specificity/resolution in k-mer space requires additional space in memory.
This problem is a good introduction to other concepts used in bioinformatics. It should be easier than alignment to teach to beginner students, especially with confusion resulting from the emphasis on nucleic acids with hype over the Illumina technology. For example, the human genome was solved by a HamiltonianIn this simple example, there is only one path through the de Bruijn graph walk through an overlap-consensus assembly graph in the year 2003, which seems like a very topical, influential, and important introductory lesson for new students, who might not be armed with the amount of computer science and coding experience to focus on dynamic programming in their first year coding in graduate school.
K-mers are also a natural segue to modern Illumina data and associated de Bruijn graphIn this example, the first image shows a graph with multiple paths from left to right. Which one is the correct one? That is part of the challenge here. assembly algorithms. While effective and powerful, processing dozens of gigabytes of raw data and requiring tens of gigabytes to store the graph in memory, there are no automatic solutions to assembly problems. Here I would introduce the concept of misassembly as crucial for students moving into that side of bioinformatics education early with respect to their CS, Bio, and Chem fundamentals.
Alignment theory is being prioritized in a nucleotide-centric technological and market environment, even though k-mer spectra are intuitive abstractions to biologists familiar with spectroscopy. Biologists must review phylogeny, genetics, then computer science, software development, bioinformatics, basic alignment and multiple sequence alignment to come full circle to the phylogenetic fundamentals they were exposed to in introductory biology. The area where perhaps biologists stand the most to gain from formal collaboration and academic exchanges are at the intersection of biotechnological instrumentation and hardware economics, given the radically permissive software development counter-culture. This would necessitate some exposure to the economics of instrumentation which may not make its way into the bioinformatics sequencing cores. In short, the educational programs need to adapt to the market of the current instrumentation and theoretical limitations, but not driven towards it for both sides of entry into the interdisciplinary system.
The current meme is that every bioinformatician will have written their own alignment algorithm by 2020. But the goal of educating the biologists is not to necessarily make each graduate competitive as software engineers when compared to the computer scientists going into bioinformatics to learn the biology; instead, the goal is to give the biologists a flavor of bad algorithm design or prioritizations where they are needed to refocus efforts on performance metrics, accuracy benchmarks, and inter-institutitional reproducibility efforts. Similarly, for biologists who are interested in the theoretical or mathematical biology of algorithm design, the focus should be on sufficient prototyping and collaborative software development, to prepare them to collaborate on advanced parallel or GPU implementations of their theories.
Sadly, I'll admit it... I don't have a good perspective on what the computer scientists need to learn necessarily from biology in order to create reasonable software that is both exciting and measurably performant, but that also may have metrics that don't look as cut and dry as reads-per-minute. /rant.
True paradigm shifts and marketing language both require experience as well as technical vocabulary to decipher. While computer scientists may observe algorithmic paradigm shifts, biologists may drive projects and complete literature review by appreciating the wealth of information available to them. Both 'styles' of bioinformaticians should be involved in biological knowledge management as well as methods development to critique and compare performance between sequencing centers in model organisms. The latter is closer in my view to applied biology, whereas algorithm focus is much more like mathematical biology. Applied biology has significant market value and will drive the establishment of basic adoption of bioinformaticians into properly integrated teams, instead of being relegated to software development positions away from the biologists who will provide them with interesting biological datasets, application ideas, and audiences for their software.
What misassemblies are and why you should care
When is a genome assembly project actually over? We are told the process ends when repetitive sequences impede graph collapse, a complete-ish picture of the contigs and their genes can be ascertained before the process produces one single perfectly linear sequence, and similarly qualitative statements about completeness in the context of biology. "Good enough" may be an easy hand-wave to pull off if you're a biologist, but if you're a computer scientist turned bioinformatician, you'll want to present as much information as possible about the biology that is present, and describe with often abused metrics (average contig length, etc) how the assembly appears to be complete. Of course, the next level for the biologists requesting the assembly is the ability to participate or re-witness the assembly process through interactivity with assembled contiguous sequences. The closest application to facilitate such exploration is the 'genome browser' and this happened to be what I built for my thesis project.
My principle software challenge during grad-school was building a resource for genomic and transcriptomic information, multiple expression profiles, transcript boundaries, genomic features, and other information considered relevant. The most challenging question related to my graduation timing was the qualitative presence of misassemblies in the final dataset; in other words, "were some of the transcript boundaries being misrepresented by artifacts introduced during the fairly automated assembly process?"
The layman can imagine that a network (graph) of these k-mers and their neighbors can fail to resolve when either too many of them stick together or vice versa. Too many of the k-mers assembling can result in misassembled contigs that should be split into one or more smaller fragments, and are thus 'artificially elongated.' On the other hand, not enough assembly would result in artificially shortened contigs, due to low local complexity in the k-mer graph.
To address this issue, I manually annotated the transcriptome assembly throughout the 3Mbp genome of the model Clostridium while integrating predicted promoter, terminator, and Shine-Dalgarno motifs, as well as known open-reading frames (ORF or CDS for coding sequence). When the occassional transcript was truncated or extended in a way that was discordant with those annotations, I corrected them. The process is manual and time consuming and after some review, I found myself in the market for an interactive assembly experience to show the students in my lab what the algorithm did automatically and why it failed just enough to need me to spot check its work. Or similarly, I would have settled for some metrics that describe the amount or extent of misassemblies present in Trinity's transcriptome assembly.
While the manual curation of >3K genes was difficult, I found the experience truly enlightening about how complete in scope some data collection or interpretation projects can be. Also, after diving that deep into assembly projects, I was surprised that there were few solutions to detecting misassemblies in an automated manner. Admittedly, the type of program has only become accessible recently due to resource constraints on consumer hardware due to random access memory (RAM) economics. I was in search of a program or metric that could be used to discover misassemblies in newly assembled contigs with respect to the reads used to construct them. This lead me to look closer at how assembly algorithms actually work, should I be put in the position to defend my use of those algorithms versus experts in the field of assembly algorithms and their shortcomings!
Okay, so what do k-mers have to do with misassemblies again?
There are many practical causes of misassemblies and imperfect reference sequences. Chimeras A genetic chimera results from rearrangements, natural or otherwise., cloning errors
A genetic chimera results from rearrangements, natural or otherwise., cloning errors Policies about genetic information are driven by rational fears, data standards, encryption, and public health concerns., transposons
Policies about genetic information are driven by rational fears, data standards, encryption, and public health concerns., transposons Transposons are cometimes referred to as 'jumping genes'., copy numbers
Transposons are cometimes referred to as 'jumping genes'., copy numbers CNV modify the numbers of a gene or genomic fragment, clonally. This can begin paralogical mutational differentiation, where useful genes are duplicated and change in sequence and, at times, purpose. Short term effects of CNVs are poorly understood, but have been implicated in certain types of cancers, as well as other diseases such as Prader-Will and Angelman syndrome./overrepresentation, and sequencing errors are symptomatic of understudied or overly complex genomes. K-mers form the basis of modern de-Brujin graph assembly algorithms (e.g. Velvet, Trinity, ABySS, SOAPdenovo) and their strategy has evolved from overlap-layout consensus (e.g. Celera, Arachne, Phrap, Newbler). To understand how misassemblies occur, one must first understand how assembly algorithms taken millions of short reads and build consensus sequences with some fundamental strategies from graph theory.
CNV modify the numbers of a gene or genomic fragment, clonally. This can begin paralogical mutational differentiation, where useful genes are duplicated and change in sequence and, at times, purpose. Short term effects of CNVs are poorly understood, but have been implicated in certain types of cancers, as well as other diseases such as Prader-Will and Angelman syndrome./overrepresentation, and sequencing errors are symptomatic of understudied or overly complex genomes. K-mers form the basis of modern de-Brujin graph assembly algorithms (e.g. Velvet, Trinity, ABySS, SOAPdenovo) and their strategy has evolved from overlap-layout consensus (e.g. Celera, Arachne, Phrap, Newbler). To understand how misassemblies occur, one must first understand how assembly algorithms taken millions of short reads and build consensus sequences with some fundamental strategies from graph theory.
Moreover, misassemblies aren't an issue faced by the human genome research community nowadays. Assembly provided the big ask to the human research community during the human genome project and then has been largely ignored in favor of mutational characterization or gene expression studies. But the best take-home lesson here is that assembled Fasta files require perhaps 10-100Mb to store, Fastq datasets can require tens of Gigabytes to store, even while compressed! There is considerable space savings on hard-drives, SSDs, flash drives and other data storage media when considering how many reads may be sequenced to provide enough redundancy and overlap to complete the assembly with guarantees of uniform coverage and sufficient depth across the contigs.
So what would you build if you could?
Large sequencing datasets take processing power and time to decompress. Storing error corrected sequence reads or k-mers as deduplicated abundances can provide significant precalculation advantages for algorithms. As an interesting note, subsampling at this stage was shown to cause some problems in downstream applications that depend on complete k-mer profiles.
Regardless, k-mer profiles and de Brujin graphs could be stored cheaply and retrieved as necessary to iterate on assemblies, k-mer partitioning, or even for interactive assembly visualizations. Read deduplication is typically thought of as something that must be duplicated code within each assembly algorithm, instead of something that is part of every pre-processing experience, graphical or otherwise. Iterating on assembly strategies would occur faster if a standardized format could be developed to store the deduplicated subsequences (reads or k-mers) and provide rapid access and annotation.
Ideally, assembly projects should be revisited with new computational methods, hardware, and datasets to look at what is discoverable today and how expensive the process truly is vs even 10 years ago. This necessitates a useful format that permits rapid assembly and quality control. Any file or database of sequencing data or their derivatives (i.e. k-mers) should also include tools for parsing, writing, and querying the data. Going back to the fastq files is certainly an option. But storage of abundances and k-mers would be orders of magnitude cheaper long term, and could pave the way for other exploratory metrics to be developed on the graphs before the assembly step.
We've covered some fundamental reasons why k-mers are important to genomic projects. Next, I'll review the existing k-mer tools.
What k-mer libraries/suites/packages are in common use?
Khmer
The Khmer package is a probabilistic approach to k-mer counting of large metagenomic datasets using the 'count-min sketch.' The package was designed for fast, online counting and subsampling. The method was designed with digital normalization in mind (or discrete normalization in the RNA-Seq literature).
The distinguishing feature of the Khmer count-min sketch (a cousin to the spectral Bloom filter, according to their paper) is the calculation of a minimum observed count across multiple hash tables, with hash collision likelihoods slightly different between the hash tables. The minimum is used to safeguard against those collisions.
In the original paper, the first implementation of Khmer was faster than Jellyfish and Tallymer in random access to k-mers. Gzip compression was offered.
In the scripts included with v2.1.2, the `count-median.py` script assumes that a sequence's abundance is proportional to its fragment abundances (okay) which is in turn proportional to the median of kmer abundances of those fragments. Occassionally this may be a reasonable assumption to make, except when bigcount is turned off, or when considering sequences/transcripts/genes with paralogous subsequences that would skew the kmer count distribution.
The v2 Khmer package offers both scripts and performant C libraries for interacting with count estimations. Many functionalities are provided in this package, and the documentation and separation into individual scripts may provide confusion to some newcomers to the bioinformatics field.
kPAL
The k-mer Profile Analysis Library was designed with inter-profile similarities in mind, with a method for normalizing against library size, and assisting researchers during modality analyses of k-mer spectra. The primary distance metrics described are a normalized Euclidean distance and what is referred to as a multiset distance.
Anvar et al. use kPAL to assess RNA-Seq, WGS, WES, and microbiome metagenomic datasets looking for "technical and biological variation present in each set of NGS data."
The 'scaling' function described in the original paper may prove useful in applications where multiple spectra are analyzed simultaneously, but might not save much time in implementations where fast profile generation is possible. The scaling and smoothing functions are more theoretically interesting. The scaling method is a type of normalization, and it would be interesting to see additional discussion in the literature of the merits of shrinking or amplifying discrete k-mer counts and the effect it has on the count distribution. While permissible, I would argue that normalization methods be done *during* distance/similarity metric calculations, and raw k-mer counts be stored with the profiles for the sake of preserving the original spectra. The smoothing function relies on a user-defined threshold for splitting larger k-mers to smooth the curve of course. While graphically this may seem to be a useful method, any smoothing function applied to datasets are transient and shouldn't be part of the *de facto* profile, but an operation applied analytically. Assembly or other analyses reliant on the original connectivity or frequencies could have an effect on their sensitivity.
However, the authors thought of many simple functions that other analysts could find useful during the interpretative process such as nullomer count (the number of 0-count k-mers), distribution generation, total counts, and strand balancing information (where applicable). The focus on simple questions makes the suite more intuitive to beginners and provides valuable information without needing to generate your own script to interogate the profile.
Another interesting feature of kPAL is the focus on both single and multiple profiles simultaneously. Metagenomics bioinformaticians may want to generate simulated metagenomes (e.g. microbiomes, ecological simulations) by combining sequencing files or profiles to create 'composite' profiles or sequencing files with species proportions known *a priori*. The inclusion of operations on arbitrary numbers of profiles is important for one of the fastest growing application areas for k-mer programs.
With respect to the choice of k:
The optimal value for *k* is the one that best separates metagenomes from randomly permuted sets.
The method they used to analytically assess their software was to generate 3 simulated metagenomes and compare them to 10 randomly shuffled metagenomic profiles (identical k-mers, different counts).
A low amount of variation in distance between the *k*-mer profiles of metagenomes and their permuted sets indicates that the distance measure... only changes according to *k*.
In other words, *k* is chosen to maximize pairwise differences between the simulated metagenome(s) and each of 10 randomly shuffled profiles. With respect to the 10 shufflings, k=10 was a minimum choice of k that sufficiently distinguishes the metagenomes from the shuffled profiles. It would be interesting to see the same figure (*mean* inter-profile distance vs k) redone for different shuffles, metagenomes, k, and even different k-mer libraries.
Anecdotally, they note that a unimodal count distribution followed from the optimal choice of k, which is preferable to having bimodal or multimodal count distributions, a hallmark of contamination, low complexity, or other confounding genomic factors. Next, they reference the same deviations from clean unimodality in the count distribution in their analysis of WES vs reference genome/transcriptome profiles.
Jellyfish
Jellyfish is the oldest and perhaps best known k-mer counting algorithm. Part of its legacy as the standard and best k-mer counter to date is of course its use of lock-free data structures for concurrent access to the k-mer counts after profile construction. In addition to the unique data structures is an encoding of the k-mer as an integer between 0 and 4k - 1.
The paper sets standards for performance analysis of almost any type of program. Memory and run-time is charted for multiple algorithms and parameterizations against the degree of subsampling for a fixed choice of k.
While very performant and strong from a computer science perspective, the article featured in bioinformatics features figures that mostly compare performance of the algorithm to other leading k-mer suites of the time, establishing the power of the implementation, but perhaps lacking on the bioinformatics side of looking at the effects of error rates, sequencing complexity, and other technical or biological artifacts on k-mer profile utility. Additionally, the CLI has a focus on profile construction, count histogram, and bloom filter with little other tools to assist in the steps of using or interpreting k-mer profiles generated by the tool.
What are key considerations for yet another k-mer suite?
-
Indexed k-mer access
The first concern with k-mer complexity is the exponent in k. Fairly reasonable choices of k (18-30bp) can cause memory issues for scripting languages; for example, Python's int type is 24 bytes. Imagine storing 1.649 Tb worth of k-mer counts in memory (that's just the counts!).
For practical reasons, access to that many k-mers and any associated graph metadata should take place with indexed file access to say the least. After generating a database of k-mer abundances, much like Jellyfish, accessing those abundances rapidly through an index file would make building assembly algorithms a breeze, saving programmers from duplicating the read deduplication and k-mer abundance calculations.
However, compression is frankly the last concern of a development process including a k-mer database, although I imagine bgzf would be attractive if the blocks could be rewritten occassionally to annotate the reads with graph properties.
-
Decoupling from assembly algorithms
Again, this is a redundant part of the assembly algorithm process, and doesn't expose the graph technologies to those that could make quality control metrics directly from assembly graphs, or create graphical representations from the actual assembly process to highlight difficult contig bridging problems.
Decoupling the read deduplication and abundance calculation steps from each assembly algorithm would help the field dramatically, given that the alignment problem receives far more attention in the human genomics community than the assembly problem since the completion of the human genome assembly in 2003.
-
Multiplexing support and inter-profile comparison
A few existing programs can merge or calculate similarity/distance between profiles do so in a pairwise manner as opposed to arbitrary numbers of input sequence files (both fasta and fastq). Also, most k-mer suites don't stream from AWS S3. I'd like to keep track of the number of total and unique k-mers and total number of reads from each file, which could be useful metadata to the user.
Perhaps more interesting to some users in the metagenomic community might be support for approximate demultiplexing with suspected references.
Is it appropriate to compare k-mer abundances directly, or would they need to be casted to more expensive floats? What is the most efficient algorithm for calculating a Euclidean distance or correlation similarity? What is the best approach to normalize against library size?
Finally, given a profile, is it possible to calculate a probability of a new sequence having been generated from the profile? Is this not a machine learning task, but a Markov-chain probability with a log-likelihood formulation and a reasonable null model? What would the null model be?
.kdb: a graph database or a simple profile file
What does the current prototype look like?
Ideally, I would like to store both the overlap graph and the k-mer profile in a file. A compressed file with an index similar to the SAM/BAM spec if possible. I'm interested in generating k-mer profiles, describing the distributions of discrete k-mer counts, and using profile similarity to recreate phylogeny and compare distance metrics in phylogenetic contexts, perhaps writing a formulation for a Markov-chain probability, and creating something that could be used to assess, anotate, and expose deBrujin graph network topology through programmatic or REST APIs.
Returning to the SAM/BAM hoisting concept... of course I like how records are a single line, but what I was most interested in an easy to read header to capture high-level statistics that you would calculate about single files during a profile's generation. Especially during an iterative process like assemblies or simulated metagenomes. An assembly algorithms could conceivably be built on top of a k-mer database that retained read information as metadata and would require less IO and storage space and hopefully less memory as a result. Metadata matters to bioinformaticians to take fastq files back to trimming, quality, or resequencing steps necessitates the most relevant statistics about the k-mers in a file.
Of course the profile is a histogram by default and any tools to provide that graph directly would be beneficial. In fact... after ommitting the YAML header in the first block... the file format for the kdb is essentially a count histogram! There is a json metadata field in the third column that could include other statistics, the connectivity of that k-mer, and other tags or numerical annotations about the k-mers role in the graph.
The .bgzf file spec provides random-ish access to rows in the file, but doesn't yet have a wrapper class for the index and file access that behaves like a dictionary given a numerical k-mer id yet. The k-mer id was borrowed directly from kPAL (above) to encode the bases in a 2bit system, such that the id is just an integer between 0 to 4k - 1. It's literally lifted directly from kPAL, and its the optimal encoding. The accessing the file through the index with the wrapper class would yield a k-mer node in the deBrujin graph, that has observed k-mer count, metadata, and connectivity information.
Is this just a histogram with random access, or is it a graph traversal tool?
It is 100% a histogram with random access currently, mostly because k-mer databases take a prohibitive amount of memory for static storage, and iteration over a file through an index would be simple to implement and demonstrate the future directions students would prefer to use a single k-mer database as part of larger application layers.
But this is how I think about what a k-mer database would need in terms of convenience functions in its API to make the assembly algorithm space more constructive, educational, and interesting for students entering that area. Imagine starting at a node and asking what the longest unambiguous path through the graph is, and what node it ends with? Should these nodes be 'tagged' in the file to unite them as a group, and that tag written back to disk? What if the intermediary nodes have not been instantiated?
The sort order in the file at the moment is just the ascending order of the k-mer id and there isn't another way to sort the nodes, yet. In that case, the range of first-last node in the unambiguous path is sufficient to tag nodes i-j, but there isn't currently a annotation class that can load nodes i-j parsimoniously into memory, tag them, and rewrite them to the file. In fact, that operation isn't supported officially by the bgzf specification directly. The hacky solution could be somewhat useful long term, but would be a beast of a coding adventure to build a prototype in Python, that wouldn't likely be rolled into production. In fact, if the project ever got that far to where re-indexing or re-writing needs required performance that couldn't manage the implementation bottlenecks, it should really just be moved to an existing database technology like sqllite3.
Moreover, the tags and other properties of a single row/kmer in the file aren't yet columns, only the k-mer id and its count are, instead the neighbor information and other metadata about the kmer are instead properties of a JSON 'metadata' structure in the 3rd column.
An aside for those unfamiliar with the BAM spec and random access to rows in the file: what do I mean by 'parsimoniously' loading a range into memory? How is that different from the generic wrapper class for the index+file to yield single nodes? Well, you wouldn't want to use the index to get the row offset, go to a block, go to the offset of that row within the block, load the node, and then return. Because then you'd have to go from the beginning of the file again to the same block, the new row's offset in the block, and then return again. You'd want the program to go to the block, read as many nodes as necessary from that block, instantiate all those nodes, and then go to the next block, read what is necessary for that block, and repeat until the final block, where you read just to the j'th line. It could just be called kdb.load_nodes(x, y) vs kdb.load_node for the Rubyists out there and if there were stylistic opinions 'load_range', kdb[x:y] or something similar could be arranged (no pun).
Now, imagine if the question was instead, what is the 'nearest' ambiguous walk that needs to be decided? This is a much harder question for me and answering requires some study of the way deBrujin or other graph assembly methods provide traversal. Even though this isn't even researched at this point, the natural question is should the categories of walks and their associated node ranges be global metadata properties stored in the header? If that is desirable, I'd need to clarify whether the routine to generate the walks should be associated with kdb.fileutil.KDB (the current, fully in-memory interface, a poorly written wrapper class around an array.array 'profile' attribute) or the (kdb, kdbfile) interface described above, which prescribes that the file is already written, and is obviously more IO intensive.
I'd make the case that for now, the in-memory size of the profile and the empty metadata array is conservatively << 400 Mb (I looked at htop when running on a Horse fecal metagenome fastq file of 15-22Gb uncompressed, 4.5-5.3Gb compressed, PRJNA590977 with a lower k-mer choice of 12), since it hasn't been populated per-node with actual metadata (tags, bools, ints, or floats). Those are things that could cause memory issues in the future, but that would require any type of input from the users on how it wants to annotate nodes as well as annotation classes operating in-memory(current) or on-disk (above). But, I haven't looked at how the file size or profile generation time scales with k or reads yet. For those who don't know, the specificity of k=12 is ~ 16M-1, which might? be enough spectral resolution for bacteria w.r.t. recreating phylogenetic distances, and might be sufficient for yeast (12M basepairs compared with 16M 12-mers), but probably isn't enough for yeast at a per-strain level to provide enough distance in profile to resolve out sequencing errors vs genuine strain differences. Again, it kind of depends on the application area, which is exactly why this is pre-alpha.
How would you gain appropriate resolution for 'interesting' applications?
Increase k. No seriously, the concept is that simple; the hardware and economics is not simple. You could likely run this thing on a c5.4xlarge (16core, 32Gb RAM) and it won't bump against memory issues in profile generation or even metadata issues yet. The big downside to the software pre-alpha is that there isn't any multiprocessing yet (there is in 2025). You could split into smaller files and merge files if you have someone who knows how to do that, and there are programmatic interfaces for that (kdb.add_profile), somewhat similar to 'merge' profile, and I would just suggest those users manually merge the header metadata from the split files. There isn't a command-line option (-p) to throw multithreading at the problem yet.
But it's Python. Why not C/C++ or something anyone would take seriously?
I've thought about answering this question more directly, but my single-developer economics prohibits me from launching several nodes (let alone spot) and babysitting them while profiling memory or single-threaded/processor performance. Plus, doesn't the GIL prohibit parallel IO? It's obvious to me with the file size that profile generation is IO bound. Annotation on-disk would similarly be IO bound. If I think users, whose demographics I don't yet know, are going to be generating tons of single-organism profiles or merged profiles to attempt to deconvolve them (i.e. phylogenetic partitioning), I could optimize the profile generation time early if I knew there'd be any 'buy in' to that specific idea. And no I don't mean watching my google analytics. I'd need someone to say 'hey this is a cool idea' and provide feedback and visibility on my associated Reddit account for my OSS development (/u/SomePersonWithAFace). I am thinking about performance at this point, but I'll highlight my plans in a future blog post about the specific and particular tuning topics as they come and go. Lifting the encoding from kPAL was the important step towards getting the current in-memory performance. I don't think I need specific help on that topic as it relates to IO, I just need to spend more time thinking about the topic.
Fast random access to the graph nodes could be possible through the use of an bgzf index file if it is difficult to keep the entire graph database in memory. Or, would it be simpler to provide conversion utilities to graph databases like Neo4J when searching for specific graph properties? Regardless of the implementation of the graphical component, it would still be useful to have graph querying layers on top that could provide graph traversal/collapse or querying capabilities.
Questions
- What is the appropriate distribution for k-mer counts?
- Does the sparseness scale linearly with the choice of k?
- How does profile generation time or memory scale with choice of k, number of input reads, or size of an assembled genome?
- How do real-world profiles from Illumina data differ from ideal profiles generated from an organism's assembled genome?
- Is there separation by species of Markov-chain probabilities of query sequences against the profile?
- What sequencing technology support should we build into the V1 prototype?